




Introduction
La prise en charge des pneumopathies interstitielles diffuses (PID) a été transformée au cours des dernières années par des découvertes majeures sur les causes génétiques des PID, le diagnostic et le traitement des fibroses pulmonaires.
Le diagnostic et la prise en charge des PID repose sur une classification faisant porter un poids important à l’étiologie de la PID (Figure 1) (connectivite, exposition professionnelle, environnementale, …) et le diagnostic de la PID s’arrêtait jusqu’à récemment à ce point. Les découvertes génétiques récentes et les progrès thérapeutiques montrent qu’il existe un continuum entre les PID secondaires et idiopathiques et qu’il faut probablement prendre en compte d’autres données comme le caractère progressif de la PID pour classer et traiter de manière personnalisée les patients atteints de PID.
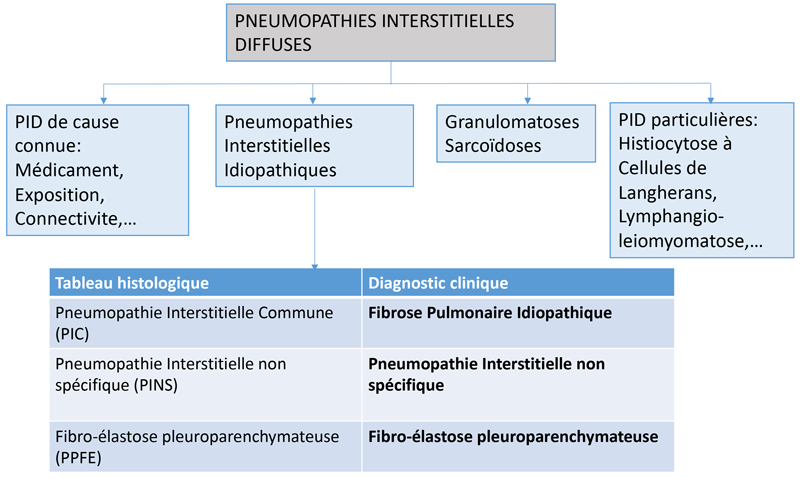
Figure 1 Classification actuelle des pneumopathies interstitielles diffuses (PID) séparant les PID secondaires des PID idiopathiques.
Génétiques des fibroses pulmonaires
Environ 10 % des patients atteints de PID ont un apparenté lui-même atteint de PID. Environ 30 % de ces formes familiales sont expliquées par une cause monogénique le plus souvent liée à une mutation d’un gène lié aux télomères. Ces mutations peuvent aussi être associées à des manifestations hématologiques (myélodysplasie, leucémie, …) et hépatiques (hyperplasie nodulaire régénérative, cirrhose, ...) qui compliquent la prise en charge et le conseil génétique de ces patients. Ainsi après transplantation pulmonaire ces patients présentent des complications hématologiques particulièrement fréquentes nécessitant l’adaptation des traitements immunosuppresseurs et cytotoxiques. De plus, alors que les traitements antifibrosant doivent être proposés chez les patients atteints de fibrose pulmonaire idiopathique (FPI, cf infra), la plus fréquente des PID idiopathique, un traitement spécifique étiologique de ces mutations pourrait être le premier traitement personnalisé des patients atteints de FPI.
En plus des mutations des TRG, d’autres variants plus fréquents ont été associés au risque de FPI. Il existe notamment un variant dans le promoteur de MUC5B, présent dans environ 10 % de la population caucasienne associé à une augmentation de la transcription de MUC5B. La présence de ce variant à l’état hétérozygote est associée à une augmentation du risque par 6 de développer une FPI. De manière intéressante, une augmentation du risque du même ordre de grandeur de PID associée à une polyarthrite rhumatoïde, d’asbestose ou de pneumopathie d’hypersensibilité (PHS) a récemment été montré[1]. Cette découverte démontre des facteurs de risques génétiques commun entre la FPI et des PID secondaires et donc un continuum entre ces pathologies.
Diagnostic des fibroses pulmonaires
Le diagnostic des PID repose sur une première étape de recherche étiologique. Vient ensuite, l’analyse de l’aspect de la PID qui va permettre de guider le traitement. En effet l’aspect de la PID est prédictif de la réponse aux immunosuppresseurs et en particulier au corticostéroïdes (Figure 2). Ainsi un aspect de pneumopathie organisée (PO) est en règle générale associé à une très bonne réponse aux corticostéroïdes. À l’inverse, un aspect de pneumopathie interstitielle commune (PIC) n’est pas associé à une amélioration fonctionnelle sous corticostéroïdes. La mise en évidence d’un aspect de PIC dans un contexte idiopathique permet le diagnostic de FPI et justifie d’un traitement antifibrosant (cf infra).
L’identification de l’aspect de PIC peut être réalisée sur l’imagerie ou sur l’histologie (biopsie pulmonaire). Les progrès du scanner et les confrontations entre les données d’imagerie et anatomopathologiques ont permis de mieux caractériser l’atteinte interstitielle vue au scanner et de limiter le recours à une biopsie pulmonaire. Il a d’abord été démontré qu’un aspect de PIC au scanner, défini par la présence d’images en rayon de miel et une topographie sous-pleurale et basale des lésions, avait une valeur prédictive positive de plus de 95 % de PIC histologique, permettant ainsi de se passer d’une confirmation histologique. Il a ensuite été montré qu’un aspect de «PIC probable» au scanner, défini par la présence de bronchectasies par traction périphériques et de réticulations, de topographie prédominante sous-pleurale et basale, sans rayon de miel, permettait également, dans un contexte clinique évocateur de FPI, de prédire une PIC histologique et ainsi de poser un diagnostic de FPI sans recours à une confirmation histologique [2].
Malgré ces progrès radiologiques permettant de diminuer le recours à une biopsie chirurgicale et en l’attente de confirmation des résultats prometteurs des analyses automatisées des scanners thoraciques, il est encore parfois nécessaire d’obtenir une confirmation histologique de PIC.
Les progrès chirurgicaux et la meilleure sélection des patients ont permis de diminuer la morbi-mortalité de cet examen invasif. Parallèlement la technique de cryobiopsie a été développée. Il s’agit d’une technique endoscopique dans laquelle une cryosonde est introduite par le canal opérateur. Cette sonde permet de congeler les tissus avoisinants sur plusieurs millimètres en quelques secondes. Un morceau de poumon gelé de 5 mm d’arête au moins peut ainsi être obtenu et permet une analyse histologique représentative de parenchyme pulmonaire. Ainsi dans une étude prospective récente, dans 95 % des cas, si le degré de confiance dans le diagnostic était élevé, le diagnostic histologique proposé par cryobiopsie était concordant avec celui de la biopsie pulmonaire chirurgicale. En revanche, quand le degré de confiance était plus faible, la classification était modifiée pour 23 % des patients entre les 2 techniques[3]. De plus, d’autres techniques encore moins invasives sont en cours de développement, et une méthode basée sur l’analyse transcriptomique des biopsies transbronchiques pour le diagnostic de FPI est ainsi déjà approuvée aux États-Unis, bien que les données soient pour l’instant encore limitées[4].
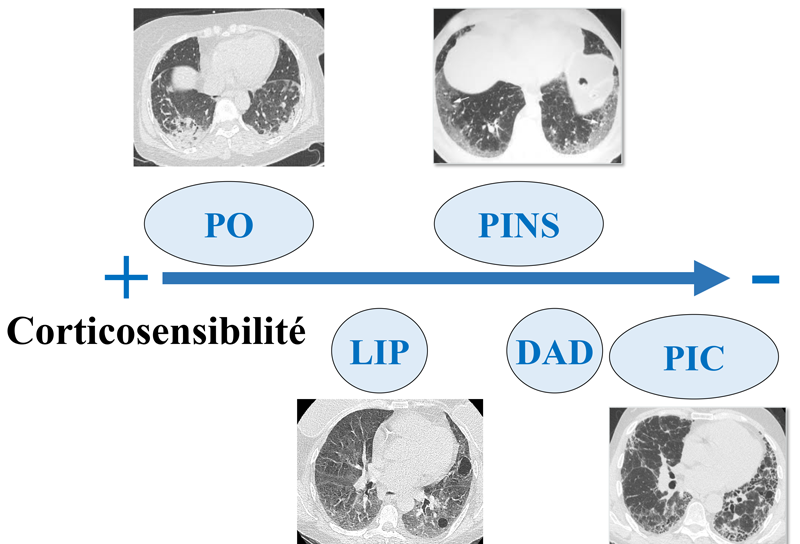
Figure 2 Corticosensibilité prévisible en fonction de l’aspect de la pneumopathie interstitielle diffuse. Pneumopathie organisée (PO), pneumopathie interstitielle lymphocytaire (LIP), Pneumopathie interstitielle non spécifique (PINS), Dommage alvéolaire diffus (DAD), Pneumopathie interstitielle commune (PIC).
Traitement des fibroses pulmonaires
Traitement de la FPI
Il a fallu attendre 2012, pour que l’inefficacité et même la nocivité d’un traitement immunosuppresseur associant azathioprine/prednisone/N Acetyl cystéine soient démontrées dans la FPI, alors qu’il était jusqu’alors recommandé[5]. Plusieurs analyses post-hoc ont suggéré que la nocivité de la trithérapie puisse être liée à des caractéristiques génétiques particulières (génotype de TOLLIP ou télomères courts), cependant il est peu probable qu’un traitement immunosuppresseur ait un effet positif dans la FPI en dehors des exacerbations ou l’indication des corticoïdes doit encore être démontrée. En effet l’étude académique prospective française EXAFIP a récemment montré que l’ajout de cyclophosphamide à la corticothérapie augmentait le risque de mortalité à 3 mois après une exacerbation de FPI (45 % vs 31 %).
Cependant, 2 traitements antifibrosants (nintedanib et pirfenidone) ont démontré leur bénéfice dans la FPI mis en évidence par un ralentissement du déclin de la CVF par rapport au placébo[6, 7]. En effet tous les patients atteints de FPI vont présenter un déclin de la CVF et il s’agit du meilleur paramètre actuellement disponible pour évaluer l’effet d’un antifibrosant. L’effet des 2 antifibrosants est sensiblement identique avec un déclin en moyenne de 240 mL/an dans les groupes placébo contre 120 mL/an dans les groupes traités par antifibrosant. La pirfenidone est ainsi disponible depuis 2012 en France et le nintedanib depuis 2016.
Les études de cohorte ont depuis rapporté une amélioration observée de la survie chez les patients ayant reçu un traitement antifibrosant[8]. L’effet sur la qualité de vie de ces traitements est cependant moins évident. En effet les effets indésirables sont importants, avec surtout une photosensibilité pour la pirfenidone (plus de 35 % des patients) et digestif pour le nintedanib (diarrhée et perte de poids pour plus de 60 % des patients). Ainsi plus de la moitié des patients arrêtent le traitement antifibrosant avant 2 ans de traitement soit en raison d’une progression de la maladie soit d’une intolérance au traitement.
Il est ainsi aujourd’hui nécessaire de disposer de nouveaux traitements antifibrosant, qui pourraient s’ajouter ou remplacer ceux actuellement disponibles en particulier en cas de mauvaise tolérance. Plusieurs molécules sont ainsi à l’étude dans des essais de phase 3 après avoir démontré des résultats prometteurs dans des essais de phase 2 comme le Pamrevlumab un anticorps monoclonal anti-CTGF. Parallèlement l’intérêt et la tolérance d’une bithérapie associant pirfenidone et nintedanib fait l’objet d’un essai clinique prospectif académique en France.
Fibroses non idiopathiques
Jusqu’à récemment le principal traitement des fibroses non idiopathiques était le traitement de la cause comme l’éviction de l’allergène en cas de PHS ou un immunosuppresseur pour les connectivites. La communauté de certains facteurs de risque (âge, genre, tabac), de l’aspect scannographique de PIC et des facteurs de risques génétiques comme MUC5B entre certaines PID secondaires (asbestose, PHS ou polyarthrite rhumatoïde-PID) et idiopathiques justifiaient d’évaluer l’effet des antifibrosants dans cette indication.
Par ailleurs le méthotrexate n’est pas un facteur de risque de PID au cours de la polyarthrite rhumatoïde et pourrait même diminuer le risque de PID alors que plusieurs études récentes ont confirmé que l’activité de la maladie évaluée par le score DAS28 par exemple était un facteur de risque de PID.
Plusieurs essais ont démontré l’intérêt du nintedanib et de la pirfenidone dans ces indications. L’essai principal (INPULSIS) nintedanib contre placébo a inclus 663 patients avec une fibrose « progressive » dont 1/4 de PHS et 1/4 de PID-connectivite incluant globalement 62 % de patients avec un aspect de PIC. Le déclin de la CVF à 52 semaines était de -80 mL dans le groupe nintedanib contre -187 mL dans le groupe placébo (p<0,001)[9].
La pirfenidone a été évaluée dans l’indication de fibrose progressive. Une première étude incluant des fibroses progressives idiopathiques n’a pas atteint le critère de jugement principal, le déclin de la CV mesuré par un spiromètre portable à domicile. En revanche, le ralentissement du déclin de la CV était statistiquement significatif quand mesuré au laboratoire d’EFR[10]. Une deuxième étude prospective arrêtée précocement a inclus 127 patients avec fibroses progressives. À 48 semaines le déclin de la CVF était significativement plus faible dans le groupe traité par pirfenidone, avec une différence de 3,5 % (95 %IC 0,21 to 6,86) entre les 2 groupes, quelle que soit la cause de la PID[11].
Parallèlement, le nintedanib a aussi démontré son efficacité dans les PID associés au sclérodermie, alors que l’aspect le plus fréquent est celui de pneumopathie interstitielle non spécifique[12]. Il pourrait y avoir un effet additif sur le déclin de la CVF avec le mycophenolate mofetil (MMF). En effet le déclin moyen de la CVF était de 40 mL/an pour les patients recevant MMF et nintedanib, contre 66 mL/an pour ceux recevant uniquement du MMF, 64 mL/an uniquement du nintedanib et 120 mL/an aucun des eux[12]. Des résultats très prometteurs ont aussi été rapportés dans des études de phase 3 sur l’atteinte pulmonaire de la sclérodermie avec 2 autres immunosuppresseurs : le rituximab (anti CD20) et le tocilizumab (anti IL6) sans AMM actuellement dans cette indication. Enfin les résultats de l’étude EVERILD, une étude académique française prospective ayant comparé MMF+placébo à MMF+ rituximab dans les pneumopathies interstitielles non spécifiques devraient bientôt être disponibles.
Conclusion
La prise en charge des patients atteints de PID a donc été transformée par la démonstration d’un continuum entre les formes secondaires et idiopathiques tant sur le plan de la présentation radiologique, que de facteurs de risque génétiques et s’est confirmée par la démonstration de l’efficacité des antifibrosants. Il est probable que traitement des PID des connectivites soit à l’avenir une combinaison de traitements antifibrosants et immunosuppresseurs dont la chronologie reste à définir.
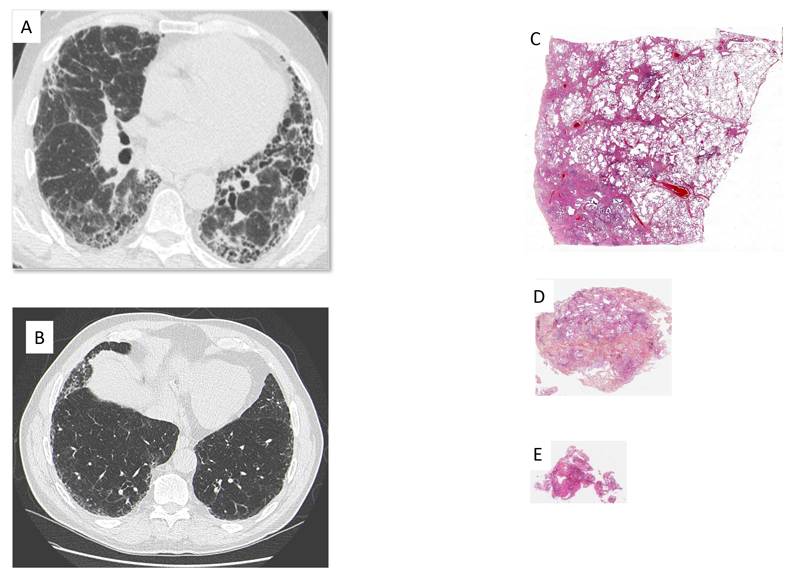
Figure 3 Scanner représentatif de (A) Pneumopathie interstitielle commune, (B) Pneumopathie interstitielle commune probable. Prélèvement histologique représentatif prélevé (A) par biopsie pulmonaire chirurgical, (B) par cryobiopsie, (C) par biopsie transbronchique.
Date de l'article : Juillet 2022
References
- Juge P-A, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, Kannengiesser C, Ottaviani S, Oka S, Tohma S, Tsuchiya N, Rojas-Serrano J, González-Pérez MI, Mejía M, Buendía-Roldán I, Falfán-Valencia R, Ambrocio-Ortiz E, Manali E, Papiris SA, Karageorgas T, Boumpas D, Antoniou K, van Moorsel CHM, van der Vis J, de Man YA, Grutters JC, Wang Y, Borie R, Wemeau-Stervinou L, Wallaert B, et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N. Engl. J. Med. 2018; 379: 2209–2219.
- Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR, Wells A, Martinez FJ, Azuma A, Bice TJ, Bouros D, Brown KK, Collard HR, Duggal A, Galvin L, Inoue Y, Jenkins RG, Johkoh T, Kazerooni EA, Kitaichi M, Knight SL, Mansour G, Nicholson AG, Pipavath SNJ, Buendía-Roldán I, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018; 198: e44–e68.
- Troy LK, Grainge C, Corte TJ, Williamson JP, Vallely MP, Cooper WA, Mahar A, Myers JL, Lai S, Mulyadi E, Torzillo PJ, Phillips MJ, Jo HE, Webster SE, Lin QT, Rhodes JE, Salamonsen M, Wrobel JP, Harris B, Don G, Wu PJC, Ng BJ, Oldmeadow C, Raghu G, Lau EMT, Cryobiopsy versus Open Lung biopsy in the Diagnosis of Interstitial lung disease alliance (COLDICE) Investigators. Diagnostic accuracy of transbronchial lung cryobiopsy for interstitial lung disease diagnosis (COLDICE): a prospective, comparative study. Lancet Respir. Med. 2020; 8: 171–181.
- Raghu G, Flaherty KR, Lederer DJ, Lynch DA, Colby TV, Myers JL, Groshong SD, Larsen BT, Chung JH, Steele MP, Benzaquen S, Calero K, Case AH, Criner GJ, Nathan SD, Rai NS, Ramaswamy M, Hagmeyer L, Davis JR, Gauhar UA, Pankratz DG, Choi Y, Huang J, Walsh PS, Neville H, Lofaro LR, Barth NM, Kennedy GC, Brown KK, Martinez FJ. Use of a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: a prospective validation study. Lancet Respir. Med. 2019; 7: 487–496.
- Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366: 1968–1977.
- Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G, Brun M, Gupta A, Juhel N, Klüglich M, du Bois RM. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011; 365: 1079–1087.
- 7. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE, Lancaster L, Sahn SA, Szwarcberg J, Valeyre D, du Bois RM, CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet Lond. Engl. 2011; 377: 1760–1769.
- Guenther A, Krauss E, Tello S, Wagner J, Paul B, Kuhn S, Maurer O, Heinemann S, Costabel U, Barbero MAN, Müller V, Bonniaud P, Vancheri C, Wells A, Vasakova M, Pesci A, Sofia M, Klepetko W, Seeger W, Drakopanagiotakis F, Crestani B. The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018; 19: 141.
- Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, Coeck C, Clerisme-Beaty E, Rosenstock B, Quaresma M, Haeufel T, Goeldner R-G, Schlenker-Herceg R, Brown KK, INBUILD Trial Investigators. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019; .
- Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, Axmann J, Kirchgaessler K-U, Cottin V. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: design of a double-blind, randomised, placebo-controlled phase II trial. BMJ Open Respir. Res. 2018; 5: e000289.
- J B, A P, M K, J J, Kf R, F B, R B, C G, M H, H W, P H, D K, S B, H W, Jh F, W N, N S, M C, N K, M F, S H, N K, S T, J F, T W, P N, A G. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. [Internet] Lancet Respir Med; 2021 [cited 2022 Feb 11]; 9Available from: https://pubmed.ncbi.nlm.nih.gov/33798455/.
- Efficacy and safety of nintedanib in patients with systemic sclerosis-associated interstitial lung disease treated with mycophenolate: a subgroup analysis of the SENSCIS trial - PubMed [Internet]. [cited 2022 Feb 11].Available from: https://pubmed.ncbi.nlm.nih.gov/33412120/.